
碳碳键(C(sp3)–C(sp3))是有机分子三维结构的核心化学键,其断裂重组反应可以实现分子结构的快速改造与重构,能为药物分子合成提供新颖、高效的合成方法。然而碳碳单键的高键能、弱极化等特性使得这类转化反应尤为挑战。特别是基于碳碳键的可逆断裂与重构碳中心实现手性富集这一课题,至今尚未得到有效解决。近日,中国科学院上海有机化学研究所金属有机化学国家重点实验室的左智伟课题组(课题组网站连接:http://zuogroup.sioc.ac.cn)利用配体金属电荷转移催化(LMCT catalysis)策略,首次实现了非张力碳碳键断裂-立体重组,为仲醇和叔醇的不对称合成提出了新范式。该催化体系建立了手性放大和手性富集的新过程,突破了去消旋化反应不能应用于连续手性和季碳手性的局限,通过系统性机理研究,进一步揭示了去消旋化反应中催化剂手性放大的倍增效应和立体选择性乘积公式(er=kRk-S/kSk-R)。相关研究成果以《Multiplicative enhancement of stereoenrichment by a single catalyst for deracemization of alcohols》为题,于2023年10月26日在线发表于《科学》杂志(DOI:10.1126/science.adj0040),中国科学院上海有机化学研究所是论文唯一通讯单位。
左智伟课题组一直致力于惰性键选择性转化研究,结合金属有机和光化学,提出并发展了LMCT催化和自由基介导惰性键均裂的协同策略(Chem Rev. 2022, 122, 2429),针对碳碳键断裂转化一般需要依赖张力释放或导向辅助活化等挑战,建立了LMCT催化烷氧自由基介导的非张力碳碳键断裂转化新体系,在过去几年中利用碳碳键作为非传统官能团实现了一系列转化反应(JACS. 2021, 143, 4896. ACIE. 2021, 60, 5370. Chem. 2020, 6, 266; JACS. 2019, 141, 10556; JACS. 2018, 140, 13580; ACIE. 2016, 55, 15319)。碳碳键断裂之后如果能够再次形成,融合LMCT催化和不对称催化模式,实现碳碳键断裂-立体重组的新过程,就能直接将消旋体转化为光学活性产品,为去消旋化提供新途径。相比于立体选择性π键光敏异构化(Bach, et al. Nature2018,564,240)和立体选择性碳氢键可逆形成为代表的策略(Knowles, et al. Science 2019, 366, 364;Luo, et al. Science 2022, 375, 869; Jiang, et al. ACIE. 2022, 61, e202211241),这一途径能够利用碳碳键的结构性优势,实现连续手性中心和季碳手性中心的去消旋化。
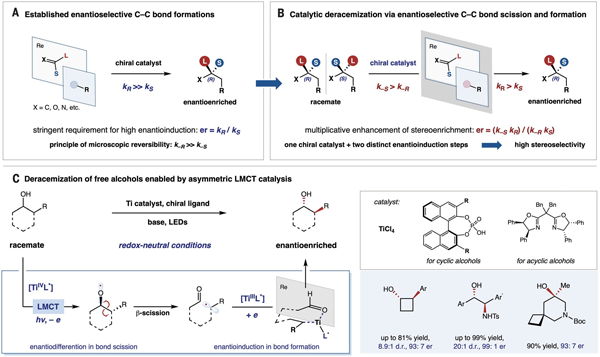
图 1. 通过对映选择性碳碳键断裂重组策略实现催化去消旋化的范例
手性醇是药物分子的重要合成砌块,虽然已有很多优异的不对称催化合成反应被报道,但是含多个手性中心的醇类化合物仍然是颇具挑战性的合成目标。考虑到消旋体可以用经典的加成反应快速合成,实现醇的去消旋化就能为这一难题提供有效的解决思路。以2-苯基环戊醇为例,该分子存在两个连续手性中心,有4个可能的立体异构体,LMCT催化去消旋化反应可以将消旋体混合物高选择性的转化为其中一个异构体(1S,2R)(图2)。在LED灯照射下, 反应仅需使用手性磷酸和钛催化剂就可以获得优异的立体选择性,不需要外加任何当量氧化还原试剂。

图 2. 反应条件优化
如图3所示,这种基于Ti-LMCT的光催化去消旋化策略适用于一系列具有连续手性中心或全取代季碳手性中心的外消旋醇。Zamifenacin,哌立度酯和贝尼地平等药物中的基本结构单元手性3-羟基哌啶也可以通过去消旋化实现手性富集。该去消旋化反应对于全取代季碳手性中心的叔醇底物同样适用,α-位被不同电性芳环或烷基取代的3-羟基哌啶叔醇可以成功实现三级碳手性中心的构建。值得一提的是,即使是高张力的环丁醇类化合物仍能很好的兼容。另外,七元环的仲醇和叔醇也能适用于去消旋化反应。
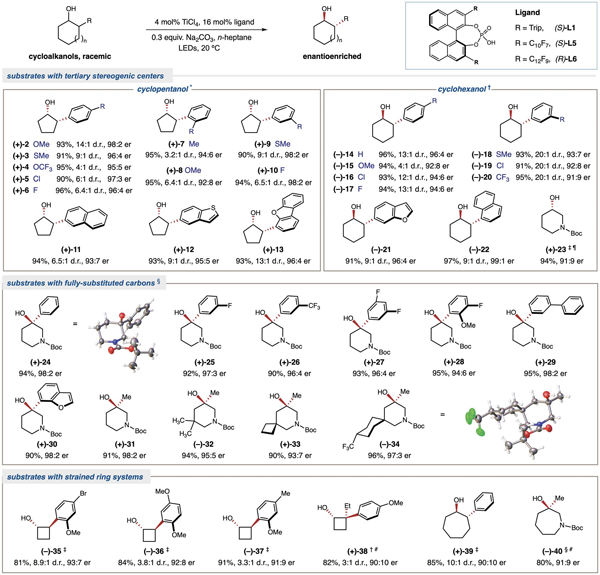
图 3. 环状醇底物范围
进一步探索发现此反应对于链状醇也同样适用。1, 2-二芳基取代的氨基醇是手性配体合成的重要砌块,传统方法路线长且难以实现高的立体选择性。当以手性双恶唑啉L8作为配体时,Ti-LMCT催化体系以优异的收率和立体选择性实现了氨基醇的消旋化,为这一类重要的手性配体砌块提供了实用的合成方法。
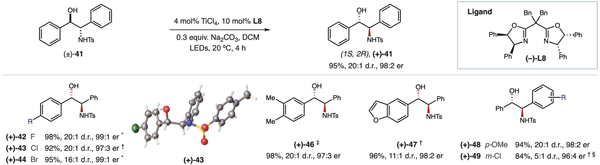
图4. 非环状醇去消旋化反应
为了探究反应可能的机理,作者首先通过氘代实验排除了HAT或串联的氧化还原过程作为可能的去消旋化途径(图5A)。使用消旋顺式、反式、或(1R, 2R)-2-苯基环戊醇,反应均以相同的立体选择性立体汇聚式的得到(1S, 2S)-构型产物,支持了反应经由C–C键断裂生成了共同的前手性中间体(图5B)。在获得[Ti(IV)L]配合物单晶(XRD 确认)的基础上,作者原位合成了配合物[Ti(IV)L(OR)],并通过1H NMR以及DOSY表征了其结构。稳态光解实验验证了Ti(IV)配合物的LMCT均裂过程(图5C)。对于环烷醇类底物14,通过EPR实验监测到由烷氧自由基快速断裂所产生的烷基自由基分别被DMPO和 PBN捕获得到的加合物的信号。为了进一步验证反应中醛作为关键中间体,作者进行了交叉实验,在氨基醇46的去消旋化体系中加入活性更高的对氟苯甲醛,新的对氟取代氨基醇的生成明确支持了醛和β-断裂产生的碳中心自由基作为去消旋化反应中的共同中间体。以上机理实验都支持反应经历了自由基介导的C–C键断裂和C–C键形成的反应途径。
最后作者对碳碳键断裂和形成步骤中的立体选择性控制和产物er值的关系进行了探讨。以氨基醇底物46为例,成键步骤的立体诱导可以通过syn-46底物的初始动力学结果估算,在反应的初始阶段,新形成anti-46的er值(75:25)可近似为成键步骤的选择性,即kR/kS = 3:1。这一结果可以进一步通过醛52和亚胺54在TiCl3, L8存在下还原偶联反应的选择性确认。接着,通过向反应体系中加入大量自由基捕获试剂55实现基于对映选择性C–C键断裂的动力学拆分,氨基醇46断键过程的选择性可估算为k-S/k-R = 8.1:1。经过简单的动力学方程推导,可以得到立体选择性乘积公式 (er = kRk-S/kSk-R),由此可以预测达到平衡时产物最终的立体选择性为96:4 er,这与去消旋化反应的实验值(er 97:3)基本一致,验证了去消旋化反应中催化剂手性放大的倍增效应。
该研究基于碳碳键的可逆断裂与重构过程实现手性富集,为手性醇的高效高立体选择性合成提供了新策略,拓展了 LMCT 催化在不对称合成中的应用,为去消旋化反应的研究提供了新思路,阐释了碳碳键选择性断裂与重组的新机制和催化剂手性放大的倍增效应,为不对称催化策略和催化剂的设计与优化提供了新思路。
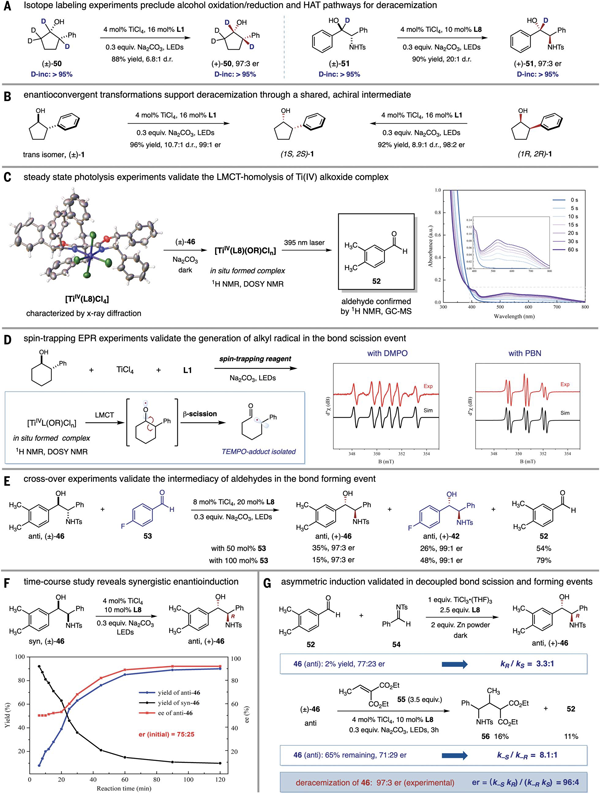
图 5. 初步反应机理研究
该论文的共同第一作者为中国科学院上海有机化学研究所金属有机化学国家重点室博士生文璐、研究助理丁佳、博士生段凌霏,左智伟研究员为通讯作者,分析测试中心吴剑博士提供了核磁共振测试方面的支持。该研究得到了国家杰出青年科学基金,科技部重点研发计划,中科院上海分院基础研究特区计划的大力资助。